
Chief Complaint: 8-year-old white female with acute awareness of complete vision loss, OS.
History of Present Illness: Patient was eating lunch on the day of presentation, when she realized that she could not see the person sitting to her left. With the assistance of the mother, they tried covering each eye alternately. The patient declared with some alarm that her entire field of view with the left eye was "completely black". The patient was seen by a local ophthalmologist and sent here for further evaluation. Upon arrival to our emergency room, the patient confirms that she "cannot see anything at all" out of her left eye. She has had no pain, no photopsias, no floaters, and no other symptoms. She does reveal that several weeks ago, another child with whom this patient had been playing told her that her left eye "looked funny" and did not seem to be "focusing on her" when they spoke.
Ocular History: Patient had routine eye examinations due to systemic risk factors (see Medical History). Prior eye examinations (including dilated fundus exam) were normal, with most recent exam at UIHC in 1998, 7 years prior to this presentation. Thereafter, patient's family had transfered eye care to local provider. Last normal exam at that location was in 2002, 3 years prior to this presentation. There is no history of eye surgery, trauma, nor contact lens use.
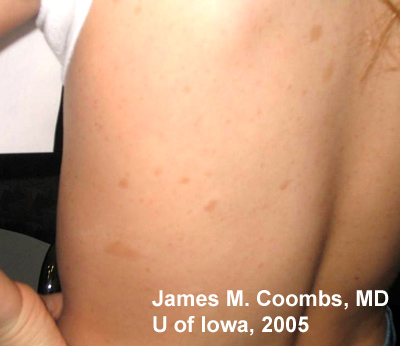
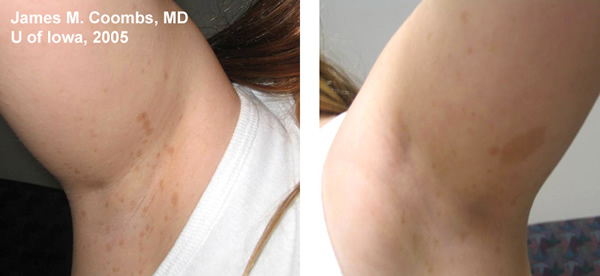
Medical History: Patient has a known history of Neurofibromatosis Type 1 (NF1 or Von Recklinghausen disease) with prior radiologic documentation of stable central nervous system (CNS) hamartomas. There is no history of seizures. There are documented skin findings consistent with NF1. Review of systems was otherwise negative.
Medications: None
Family History: Family history of Neurofitromatosis Type 1 inherited in autosomal dominant fashion through the maternal line. The mother and sister have NF1.
Social History: Active, well-adjusted elementary school student.
 |
 |
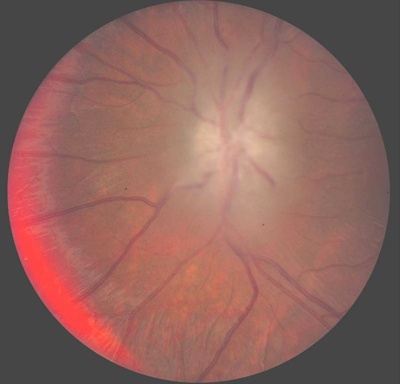 |
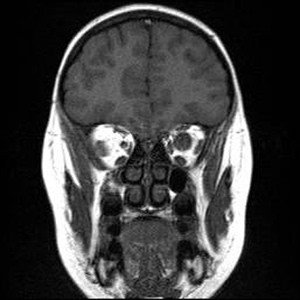
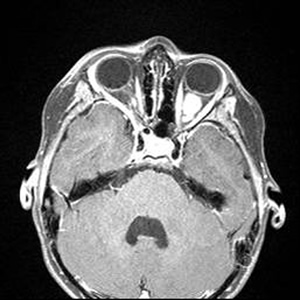
Course: In summary, this is an 8-year-old patient with a known history of NF1 now with acute awareness of complete vision loss, OS, with associated optic disc pallor and edema. Magnetic resonance imaging (MRI) of the head and orbit was obtained to investigate a possible optic nerve or intracranial tumor. A well-circumscribed, fusiform enlargement of the left optic nerve was revealed on MRI. The margins of the tumor appeared smooth and there was no calcification. The mass is isointense on T1, hyperintense on T2, and enhances with contrast (see Figures 4 and 5).
 |
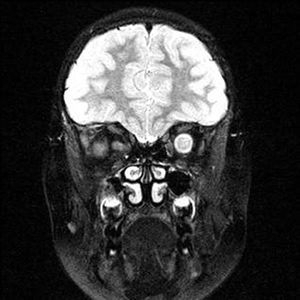 |
| 4A: MRI, T1 coronal section reveals circumscribed enlargement of the optic nerve, OS, with an isointense signal. | 4B: T2 imaging in a similar coronal plane reveals hyperintense signaling of the mass that is contained within the dura of the optic nerve, OS. |
 |
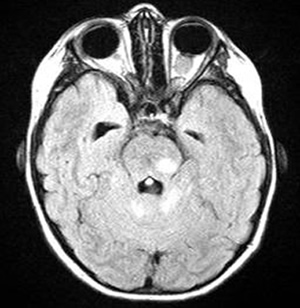 |
| 5A: This axial, post-contrast image demonstrates enhancement of the fusiform, kinked shaped optic nerve tumor. | 5B: In this axial FLAIR image, the fusiform image of the optic nerve tumor is again seen. Also here evident are several enhancing lesions in the midbrain and cerebellum, consistent with the classic hamartomatous CNS neurofibromas of NF1. |
Findings on MRI confirmed the suspicions of an optic nerve "glioma" as well as the several previously noted CNS hamartomatous lesions. Definitive confirmation of the mass as a pilocytic astrocytoma (the correct name for these "gliomas") would require biopsy and appropriate histopathology. However, the eye is already without light perception, it is non-painful, and the lesion appears to be limited to the optic nerve without intracranial extension. The family decided not to pursue a biopsy at this time. Serial imaging will be obtained in follow-up examinations to ascertain that the tumor does not extend intracranially.
Neurofbromatosis is a rare condition characterized by hamartomas of neural crest origin (phakomatosis). The condition is autosomal dominant, though spontaneous new mutations are not uncommon. The National Institutes of Health (NIH) divide neurofibromatosis into type 1 (NF1 or von Recklinghausen syndrome) and type 2 (NF2, acoustic neurofibromatosis, or central neurofibromatosis).
Only ~10% of NF patients have NF2, which is associated with bilateral acoustic neuromas/schwannomas. The defect in NF2 is on chromosome 22q. In addition to acoustic neuromas, NF2 patients frequently have ependymomas of the spinal cord or multiple meningiomas.
85% of patients with neurofibromatosis suffer from NF1, similar to the patient in this case report. The genetic defect in NF1 is almost always a mutation in the neurofibromin gene on chromosome 17 (17q11.2). Neurofibromin normally functions to down-regulate the p21 ras oncoprotein. Loss of the tumor suppresor function of neurofibromin leads to proliferation of neural tumors. The most common features of the disease include multiple neural tumors (neurofibromas) on the skin or within the body, pigmented skin lesions (ie. cafe au lait spots), and Lisch nodules (pigmented iris hamartomas).
Clinical diagnosis of NF1 requires 2 or more of the following:
The extent to which neurofibromas develop in each patient is variable, with some patients having only a few while other patients develop hundreds or even thousands of these tumors. Most commonly, patients with NF1 develop some combination of the classic findings listed in the diagnostic criteria above. However, neurofibromas may develop anywhere in the body, with varied consequence. Some patients develop spinal lesions, gastrointestinal tract tumors, osseous lesions, pancreatic involvement, vascular dysplasias, hypertension, and neurofibromas of the female genitourinary tract potentially affecting childbirth.
For the ophthalmologist, NF1 may present with a variety of ophthalmic manifestations. Lisch nodules (melanocytic nevi of the iris and uveal tract) are the most common finding. Development of a plexiform neurofibroma within the lid can cause a classic S-shaped ptosis in the pediatric patient. Optic nerve gliomas occur in 12% of patients with NF1 with most of these occuring within the first decade of life. Imaging reveals a fusiform, circumscribed enlargement of the optic nerve and nerve sheath. The tumor has no calcification, is iso- or hypointense on T1 and hyperintense on T2 MRI imaging. There is variable enhancement of the tumor with gadolinium. Imaging that exposes these optice nerve gliomas may also reveal the high signal intensity lesions in the basal ganglia, thalamus, brainstem, cerebellum, or subcortical white matter consistent with CNS hamartomas. Such was the case with this patient.
EPIDEMIOLOGY of NF1
|
OCULAR SIGNS of NF1
|
SYMPTOMS of OPTIC NERVE GLIOMA
|
TREATMENT of OPTIC NERVE GLIOMA
Guidelines for therapy include:
|
Graff JM, Coombs JM, Pramanik S. Neurofibromatosis Type 1 - Optic Nerve Glioma: 8-year-old white female with acute awareness of complete vision loss, OS. EyeRounds.org. May 15, 2005; Available from: http://www.EyeRounds.org/cases/38-NeurofibromatosisOpticNerveGlioma.htm.

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
