
INITIAL PRESENTATION
Chief Complaint: Bilateral retinal detachments
History of Present Illness:
A 48-day-old boy was referred for evaluation of bilateral retinal detachments. The pregnancy had been complicated by intrauterine growth restriction with a birth weight of 2.6kg. The infant was otherwise healthy. He was first noted to have abnormal red reflexes, non-reactive pupils, and pale discs with concern for overlying vascular abnormalities when evaluated by an outside pediatric ophthalmologist on day 8 of life. An outside retinal surgeon diagnosed bilateral, macula-involving tractional retinal detachments and performed same-day, bilateral lens-sparing vitrectomies. He was seen at the University of Iowa 1 month after surgery. Given his history, an examination under anesthesia (EUA) was performed.
Past Ocular History: Bilateral tractional retinal detachments, status post lens-sparing vitrectomies
Medical History:
Medications: Cholecalciferol
Allergies: No known allergies
Family History: Unremarkable
Social History: Unremarkable
Review of Systems: Unremarkable other than as noted in history of present illness.
OCULAR EXAMINATION
Orthophoric by Hirschberg testing with low amplitude, conjugate nystagmus in the horizontal plane.
|
Right Eye |
Left Eye |
|
|
Lids/lashes |
Normal |
Normal |
|
Conjunctiva/sclera |
Normal |
Normal |
|
Cornea |
Normal |
Normal |
|
Anterior Chamber |
Deep & quiet |
Deep & quiet |
|
Iris |
Single posterior synechia at 6 o’clock |
Normal |
|
Lens |
Clear |
Clear |
|
Anterior Vitreous |
Normal |
Normal |
|
Right Eye |
Left Eye |
|
|
Vitreous |
Clear |
Clear |
|
Disc |
Mild pallor |
Slightly elevated, mild pallor, dragged temporarily |
|
Cup-to-disc ratio |
0.1 |
0.1 |
|
Macula, vessels, and periphery |
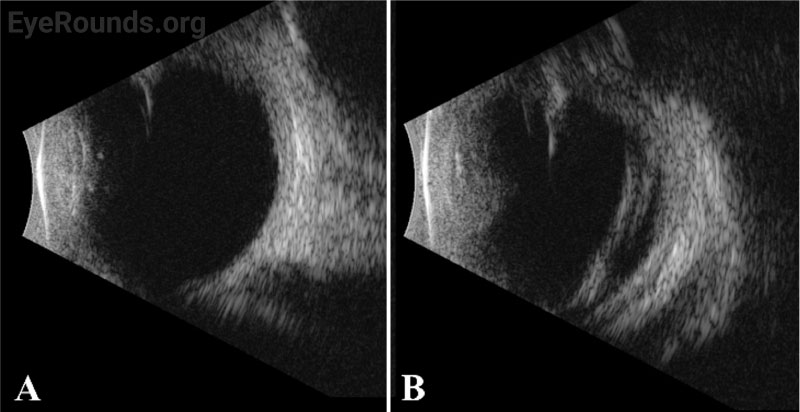
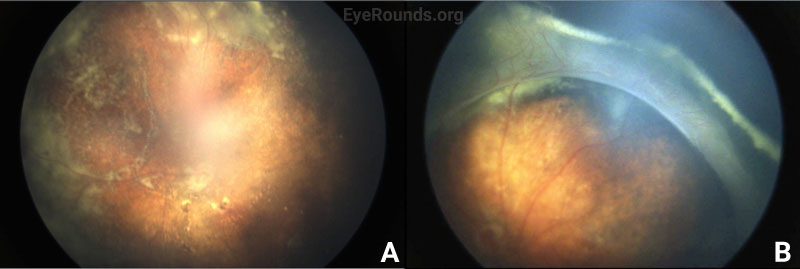
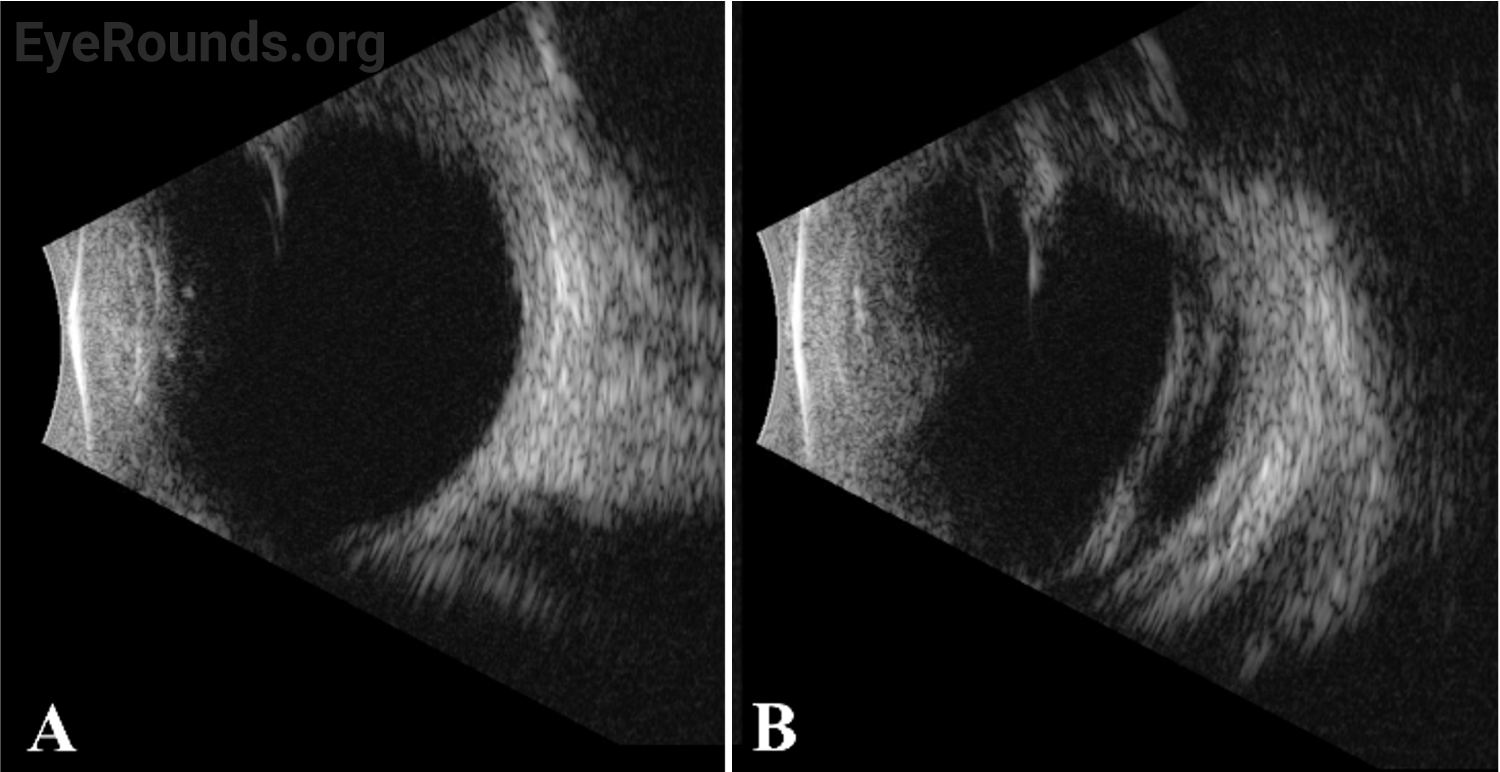
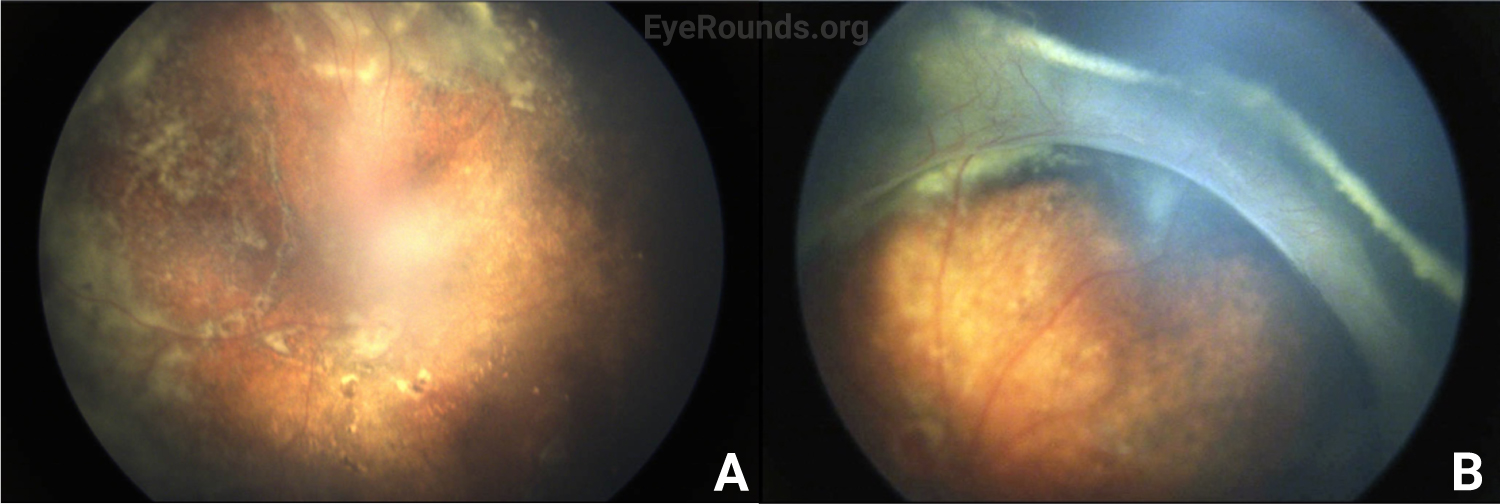
Vessel dragging/straightening; diffuse pigmentary clumps in the periphery; subretinal exudation with inferior shallow detachment |
Vessel dragging/straightening; diffuse pigmentary clumps in the periphery; less subretinal exudation than right eye concentrated peripherally; 360-degree peripheral anterior loop traction with fibrovascular proliferation, underlying vessels, and traction on peripheral retina |
Differential diagnosis:
DIAGNOSIS: Familial exudative vitreoretinopathy (FEVR)
CLINICAL COURSE
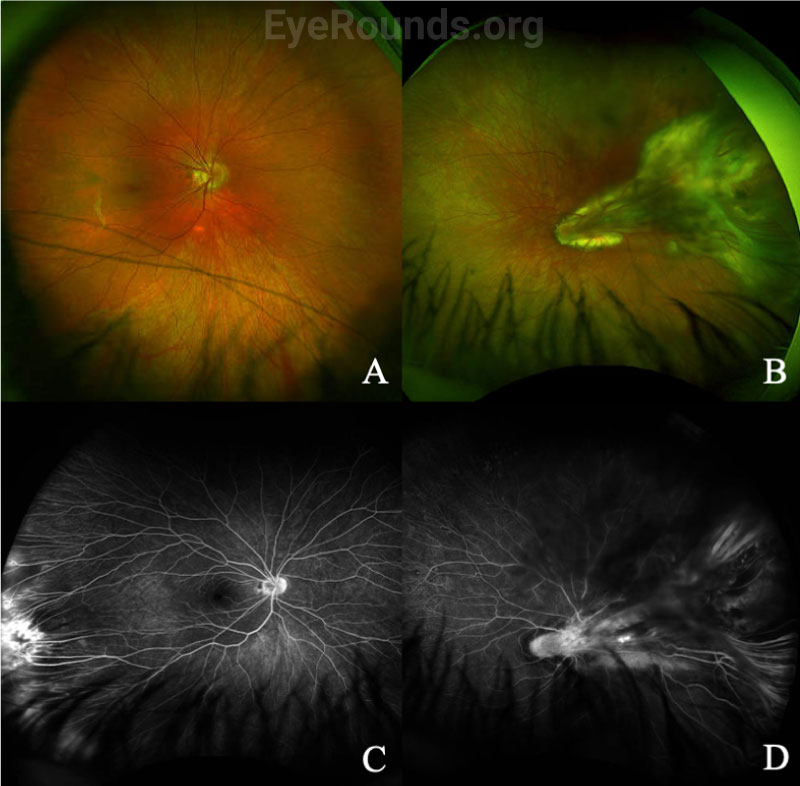
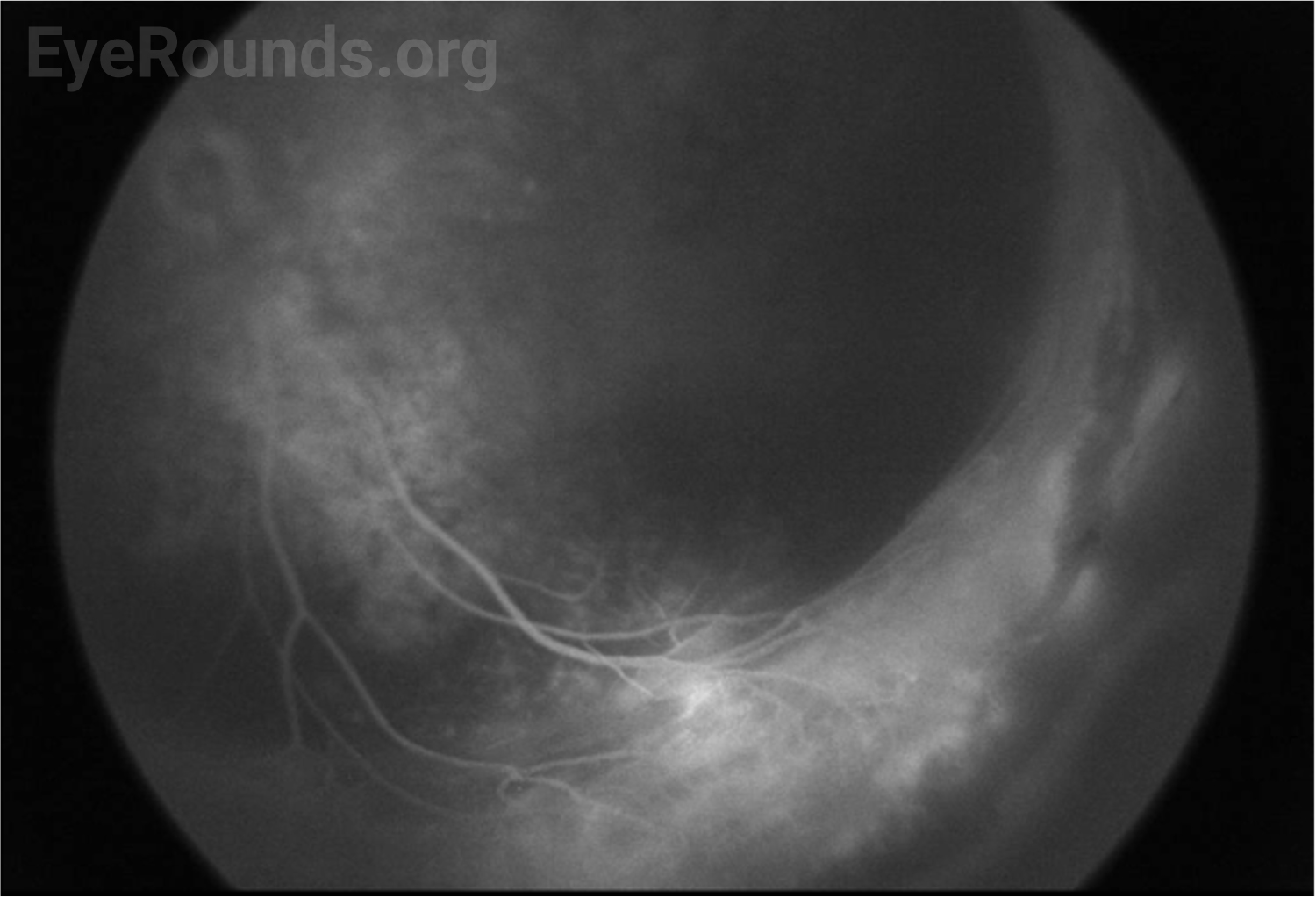
The patient’s exam findings on EUA were consistent with bilateral familial exudative vitreoretinopathy (FEVR), stage 3B.[1] Operative notes from the outside surgeon indicated that at the time of vitrectomy there was bilateral stage 4B disease. During the EUA, fluorescein angiography demonstrated peripheral neovascularization and ischemia 360 degrees extending to the posterior margin of zone 2 (Figure 3). Given the extensive bilateral neovascularization, panretinal photocoagulation (PRP) was applied to areas of ischemia, which extended from 2–3 disc diameters anterior to the macula up to the visualized fibrovascular ridge. Ablative laser was also applied to areas of shallow retinal detachment that were ischemic but without overlying fibrovascular membranes.
The patient returned one month later for post-operative evaluation. His mother reported increased responsiveness and tracking to bright light. Fundus exam showed extensive retinal scarring. The retinas were attached except for residual anterior loop traction with peripheral tractional detachment in the left eye. Six months later, the patient was tracking to light, and the retinas remained attached.
Genetic testing showed an LRP5 mutation (single copy variant of uncertain significance c.685C>T).
DISCUSSION
Etiology/Epidemiology
FEVR was first reported in 1969 by Criswick and Schepens, who described a progressive, inherited retinal vascular disease resembling retinopathy of prematurity (ROP) that occurred in patients without prematurity.[2]
Due to its rarity and heterogenous nature, the exact prevalence of FEVR is unknown. There is no gender predilection. Mutations in at least 10 genes (LRP5, FZD4, NDP, ZN408, KIF11, TSPAN12, CTNNB1, JAG1, DOCK6, and ARGHGAP31) have been associated with FEVR.[3-21] However, penetrance and expressivity are variable within families. Only 35-50% of patients with FEVR have a detectable mutation in one of these 10 known causative genes.[1]
Pathophysiology
FEVR results from mutations that disrupt signaling in the transduction pathways controlling retinal vascular development, cellular migration, and differentiation. Because of abnormal retinal angiogenesis, there can be exudative retinopathy and/or peripheral retinal ischemia, leading to neovascularization, retinal dragging, and tractional retinal detachment.
As mentioned above, FEVR is caused by mutations in multiple genes. The inheritance pattern can be autosomal dominant, autosomal recessive, or X-linked, though autosomal dominant is most common.[2,3] Most FEVR genes, including LRP5, FZD4, TSPAN12, and NDP, are involved in signal transduction within the WNT pathway, which regulates retinal vascular development.[2] Additional genes and pathways linked to FEVR are beyond the scope of this review.
Signs/Symptoms
FEVR presents with bilateral, abnormal retinal vasculogenesis in children without underlying risk factors for retinopathy of prematurity (i.e., full term, normal birth weight, no supplemental oxygen use). In children, FEVR can be diagnosed because of an abnormal newborn exam, behavioral changes, strabismus/amblyopia, or asymptomatic screening in families carrying a FEVR diagnosis. Between 10-50% of patients have a positive family history.[23] Some affected family members may be asymptomatic but have clear vascular abnormalities on fluorescein angiography.[1]
FEVR is a clinical diagnosis based on dilated fundus examination, often with supplemental fluorescein angiography. FEVR can feature some or all of the following: avascular peripheral retina (most often the temporal quadrant), dragged retinal vessels and macula (temporally), retinal falciform fold, neovascularization, vitreous hemorrhage, subretinal/intraretinal exudates (occasionally in massive quantities, resembling Coats disease), retinal detachment (which can be exudative, tractional, and/or rhegmatogenous; they occur in 21-64% of patients, and present bilaterally in 68%), persistent fetal vasculature, epiretinal membrane, and peripheral pigmentation (most prominent in the avascular retinal areas).[2,3] As a result of macular temporal dragging, patients may have pseudo-exotropia. Staging criteria for FEVR are shown in Table 1.
Stage |
Description |
Sub-stage |
|
A |
B |
||
1 |
Avascular periphery and/or anomalous intraretinal vasculature |
Without exudate |
With exudate |
2 |
Avascular periphery with preretinal neovascularization |
Without exudate |
With exudate |
3 |
Extramacular retinal detachment |
Without exudate |
With exudate |
4 |
Subtotal macula-involving retinal detachment |
Without exudate |
With exudate |
5 |
Total retinal detachment |
Open funnel |
Closed funnel |
Not all cases of FEVR are symptomatic or progressive.[4] Some patients are not diagnosed with FEVR until adolescence, when the disease often progresses. Rarely, a patient is not diagnosed until adulthood, such as if they are screened because of a FEVR diagnosis in a symptomatic relative. The average age at diagnosis is 58.6 months.[3] Severity is often inversely proportional to age of diagnosis. FEVR rarely progresses in adulthood, although vitreous hemorrhage and other vision-threatening complications can occur.[5] Thus, long term follow-up is imperative.
Testing/Laboratory work-up
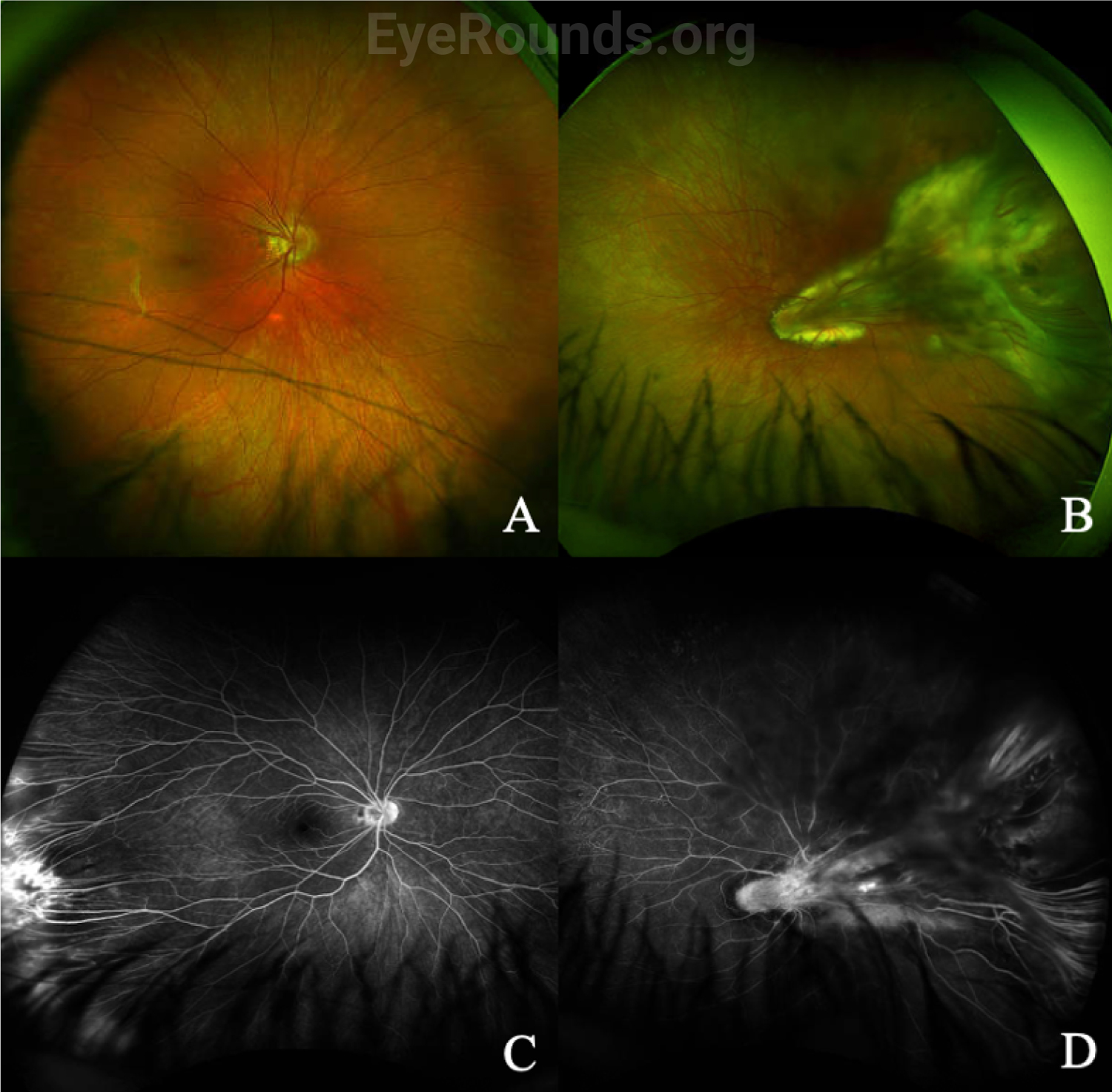
In addition to a thorough dilated fundus exam, which may require EUA in children, wide-angle fluorescein angiography (FA) is helpful in diagnosing FEVR, especially in milder cases where only the far peripheral retina is affected. Typical findings on fluorescein angiography include a V-shaped avascular zone in the temporal periphery.[1] Peripheral avascularity can extend 360 degrees in more severe cases. Additional evidence on FA that suggests FEVR includes vessel straightening, peripheral telangiectasias, neovascularization, and small-vessel fluorescein leakage, as shown for a separate patient in Figure 4.[2]
Genetic testing is important to verify the diagnosis and to counsel patients and families. However, since fewer than 50% of FEVR cases can be attributed to a known gene, a negative genetic test result should not rule out a diagnosis of FEVR.[2]
Treatment/Management
The management of FEVR depends on the stage of disease. Stage 1 disease can be observed, while Stage 2 FEVR is usually treated with laser photocoagulation of avascular zones to decrease risk of exudation and neovascular complications such as vitreous hemorrhage and retinal detachment.[2] Compared to anti-VEGF agents, laser remains the mainstay of treatment, though anti-VEGF injections are sometimes used adjunctively, especially in the presurgical period to minimize intraoperative hemorrhage.[2,3] Treatment for stage 3-5 FEVR depends on the cause of retinal detachment. If exudative in nature, laser to avascular retina can be effective. Tractional and rhegmatogenous retinal detachments usually require retinal surgery; in infants, this is usually lens-sparing vitrectomy, whereas older children can often be treated with scleral buckling.
Genetic testing results inform screening of asymptomatic family members, a small percentage of whom may require treatment. Counseling female carriers of child-bearing age can facilitate pre-implantation genetic diagnosis and/or prompt screening of their newborns.
EPIDEMIOLOGY AND ETIOLOGY
|
DIAGNOSIS
|
SIGNS/SYMPTOMS
|
TREATMENT/MANAGEMENT
|
References
Silverman JIM, Motlagh MN, Boyce TM, Russell JF. Familial Exudative Vitreoretinopathy (FEVR). EyeRounds.org. August 25, 2022. Available from https://eyerounds.org/cases/333-FEVR.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links

{kind=link}
{kind=link}
{kind=link}
{kind=link}