Chief complaint: Progressive ptosis
History of Present Illness: A 58-year-old female presents with a gradual progressive worsening of bilateral blepharoptosis. She underwent blepharoplasty to both upper eyelids seven years prior to presentation to our clinic. She also has mild dysphagia and proximal muscle weakness.
Past Ocular History: Blepharoplasty in 2003
Medical History: Non-contributory
Medications: Adderall (dextroamphetamine and amphetamine), aspirin, bupropion, vitamins, trazodone
Allergies: none
Family History: She has a brother with bilateral ptosis, dysphagia, and profound muscle weakness.
Social History: Non-smoker
Review of systems: Pertinent findings are within the History of Present Illness
Visual Acuity: 20/20 right eye (OD), 20/20 left eye (OS)
Pupils: 5 → 3 brisk, equal, no RAPD
Extraocular movements: full
Confrontation visual fields: full
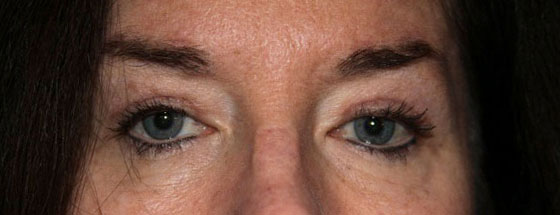
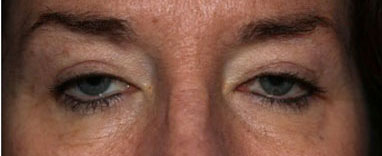
External
 |
|
 |
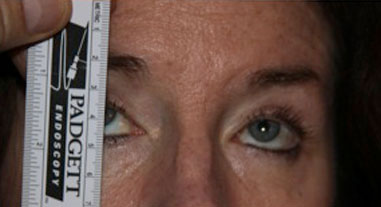 |
Dilated Fundus Exam: Normal disc, macula, vasculature, and periphery OU
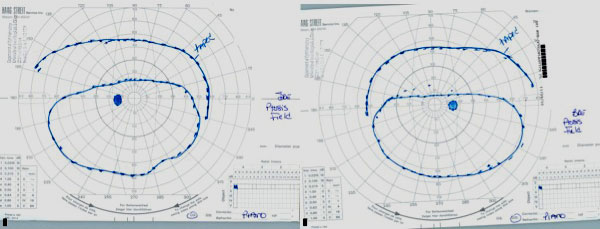
Goldmann Visual Fields: 30 degrees of improvement on the left and 39 degrees of improvement on the right, taped vs. untapped (Figure 2).
Diagnosis: Oculopharyngeal muscular dystrophy (OPMD)
|
Oculopharyngeal muscular dystrophy (OPMD) is an autosomal dominant disorder characterized by slowly progressive bilateral ptosis, dysphagia, extraocular muscle weakness, and proximal limb weakness. It was first termed OPMD by Victor and colleagues in 1962 (Victor et al., 1962). OPMD falls within the category of non-congenital, myogenic ptosis, which also includes chronic progressive external ophthalmoplegia and myotonic dystrophy (Wong et al., 2002). The largest population of patients with OPMD are those with French-Canadian ancestry who reside in Quebec (Duranceau et al., 1983). Other clusters of this disease are found in the Bukhara Jewish immigrants in Israel and individuals of Hispanic descent in the State of New Mexico (Becher et al., 2001; Blumen et al., 1997). OPMD usually starts insidiously during the fifth or sixth decade of life. These patients suffer from a progressive, bilateral, myogenic ptosis. Dysphagia is also a prominent feature of OPMD, and is initially noticed with solid foods, but can progress to difficulties swallowing liquids. Aspiration pneumonia and malnutrition are the leading cause of death in patients with OPMD, but do not shorten life expectancy because these tend to occur late in the disease. In addition to ptosis and dysphagia, patients can also have proximal limb weakness of varying severity, with some patients suffering from profound limb-girdle weakness. There is also often some element of extraocular muscle weakness (usually affecting supraduction), but complete external ophthalmoplegia is rare.
Although not well reported, patients with OPMD can also develop extremity pain and changes in their voice. Dysphonia is something that physicians who have worked with a lot of OPMD patients notice. Some claim they can diagnose the disease from the sound of the patient's voice. Arm and leg pain are symptoms that OPMD support groups note in a significant portion of their members.
OPMD is caused by an expansion of the 6 GCG trinucleotide repeat within exon 1 of the polyadenylate binding protein nuclear 1 (PABPN1) gene on chromosome 14 (Brais et al., 1998). Expansion of the GCG trinucleotide repeat to 8-13 repeats [(GCG)8-13] results in OPMD and is usually inherited in an autosomal dominant fashion with almost complete penetrance by age 70. The most common expansion is to 9 repeats, which is seen in the three large population clusters (Blumen et al., 2000). Although there is no clear relationship between the size of the repeats and the severity of the disease, the most severe phenotype is seen in patients homozygous for an autosomal dominant mutation in PABPN1 in whom the average onset of disease is 18 years earlier than in (GCG)8-13 heterozygotes (Blumen et al., 1999; Brais, 2009). Possession of the (GCG)7 repeat expansion on the other allele acts as a modifier resulting in worsening of the disease when paired with a (GCG)8-13 repeat expansion. Homozygosity for the (GCG)7 repeat expansion leads to a rarely seen autosomal recessive form of OPMD (Brais et al., 1998). Unlike other large triplet repeat expansion diseases, such as Huntington disease, PABPN1 mutations are mitotically and meiotically stable and therefore do not undergo anticipation (Brais et al., 1998).
The molecular pathogenesis behind OPMD is still uncertain. The normal PABPN1 gene encodes a protein involved in mRNA processing that is constitutively expressed within the nucleus of all cells, especially skeletal muscle (Ruegg et al., 2005). It has been hypothesized that the mutated protein in OPMD leads to the development of intranuclear inclusions and cellular toxicity through a variety of proposed mechanisms, including the disruption of the ubiquitin-proteasome pathway and the dysregulation of cellular RNA metabolism (Abu-Baker and Rouleau, 2007). The visible intranuclear inclusions seen within muscle fibers of patients with OPMD may be causative or merely a sign of toxicity of the disease (Brais, 2009).
Patients with OPMD are notoriously misdiagnosed. Probably the most common misdiagnosis is myasthenia gravis. Asking all patients who present with late onset myogenic ptosis whether they have a first degree relative with the same symptoms or problems swallowing is mandatory. Often, the swallowing difficulties are difficult to elicit early in the disease. A telltale sign of dysphagia is taking a longer time to finish meals. Though, the disease does have a world-wide distribution, it is often useful to determine whether the patient's heritage is from one of the three known geographic clusters.
Diagnosis of OPMD is based on clinical presentation, and can be confirmed by genetic testing with sequencing of the first exon of PABPN1 in order to determine the size of the GCG trinucleotide repeat. Before genetic testing was available, the diagnosis was based on muscle biopsy, which demonstrates rimmed vacuoles within the muscle fibers, small angulated fibers, and intranuclear inclusion bodies (Ruegg et al., 2005). The preferred diagnostic test is now genetic testing; muscle biopsy is no longer necessary.
Unfortunately, there is no cure for OPMD. Current therapies aim to improve symptoms that result from ptosis and dysphagia. The chosen method of ptosis repair is critical in these patients. Some advocate the use of blepharoplasty, levator advancement, or combined aponeurosis-Muller muscle advancement if the patient has adequate levator function at the time of evaluation (Burnstine and Putterman, 1999; Johnson and Kuwabara, 1974; Jordan and Addison, 1993; Kang et al., 2002; Wong et al., 2002). However, since OPMD results in progressive dystrophy of the levator muscle, many patients initially treated with levator-based surgical intervention will require repeat surgery (Allen et al., 2009). Therefore, we strongly recommend a primary frontalis suspension procedure in the majority of patients with OPMD because of the progressive nature of the disease, even if levator function is measured as "good". We prefer the use of silicone rods because it allows for postoperative adjustments and the elastic nature of silicone provides some protection against exposure keratopathy. Fortunately, patients with OPMD usually retain good orbicularis function and Bell’s phenomenon, lessening the risk of exposure keratopathy.
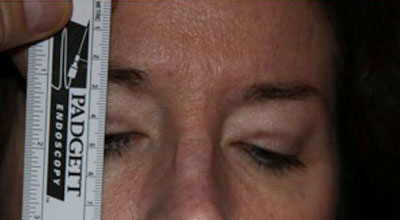
Oculopharyngeal muscular dystrophy was confirmed in our patient with genetic testing for PAPBN1, which showed one allele with the normal 6 repeats (GCG)6 and one allele with 9 repeats (GCG)9, the most common mutation associated with autosomal dominant OPMD. She had previously undergone blepharoplasty of the upper lids seven years prior to her presentation to our clinic, supporting the previous point that lid interventions are merely a temporizing measure requiring further surgery at a later date. Although her levator function was decent at 12mm, frontalis suspension is preferred over other ptosis interventions because of the knowledge that this is a progressive myogenic disease. The patient underwent frontalis suspension using a silicone sling, and her one month post-op appearance was very satisfactory (Figure 3).
|
EPIDEMIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT
|
Diagnosis confirmed by genetic testing showing expansion of the GCG trinucleotide repeat to 8 to 13 repeats within the first exon of PABPN1 gene.
Chen JJ, Allen RC. Oculopharyngeal muscular dystrophy. EyeRounds.org. January 18, 2012; Available from: https://eyerounds.org/cases/147- Oculopharyngeal-muscular-dystrophy.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links