
Chief Complaint: Progressive vision loss and glare
History of Present Illness (HPI): A 68-year-old female was referred by an outside ophthalmologist for evaluation of an anterior stromal corneal dystrophy causing progressive decline in vision with glare over the past 10 years.
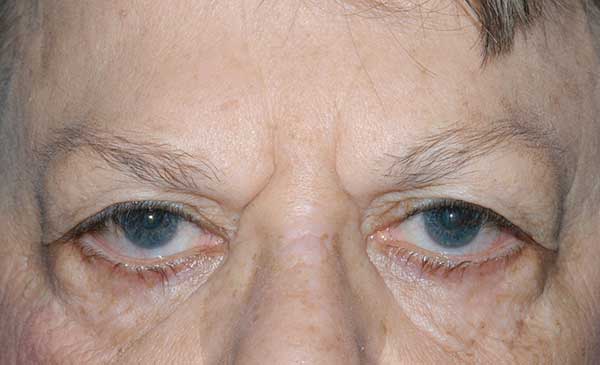
She complained of having to manually lift her upper eyelids to improve her superior visual field and had not been able to raise her eyebrows for approximately 25 years, a trait also present in both of her sisters. She was unaware if this affected either of her parents or her brother, but states that her father had an unknown problem with his corneas that caused him to have poor vision as well as droopy eyelids.
Ocular meds
Systemic meds
Allergies
Right |
Left |
|
|---|---|---|
External |
Brow ptosis |
Brow ptosis |
Exophthalmometry (Base: 84 mm) |
19 mm |
19 mm |
Cochet-Bonnet aesthesiometry |
1 cm |
1 cm |
Inferior scleral show |
3 mm |
2 mm |
Margin reflex distance 1 |
2 mm |
2 mm |
Lagophthalmos |
1 mm |
1 mm |
See Figures 1-5 below.
OD |
OS |
|
|---|---|---|
Lids/Lashes |
Dermatochalasis; poor bells; lash ptosis; prolapsed lateral fat pad; lower lid laxity, ectropion and inferior scleral show |
Dermatochalasis; poor bells; lash ptosis; prolapsed lateral fat pad; lower lid laxity, ectropion and inferior scleral show |
Conjunctiva/Sclera |
Clear and quiet |
Clear and quiet |
Cornea |
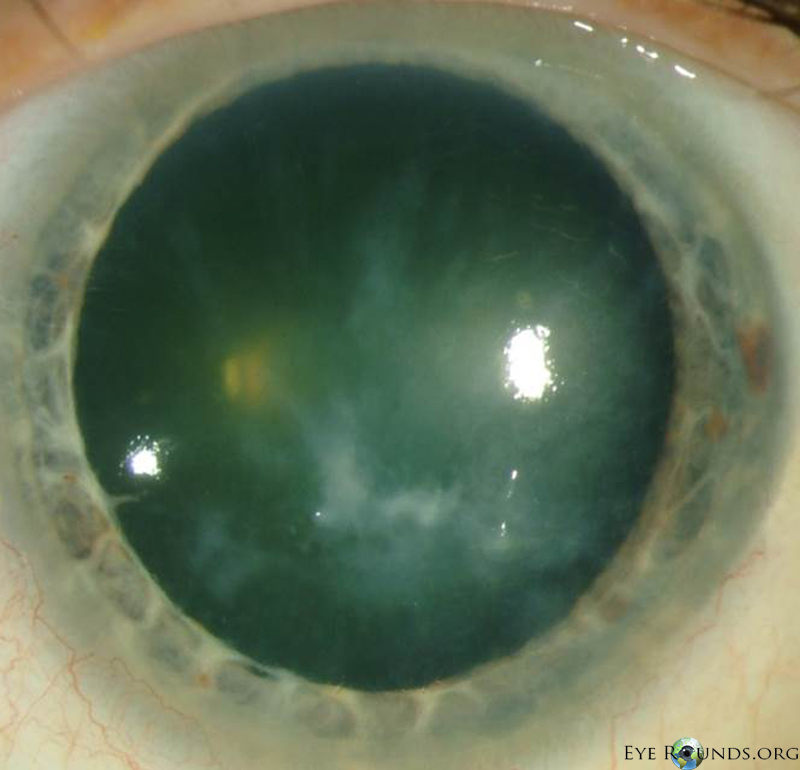
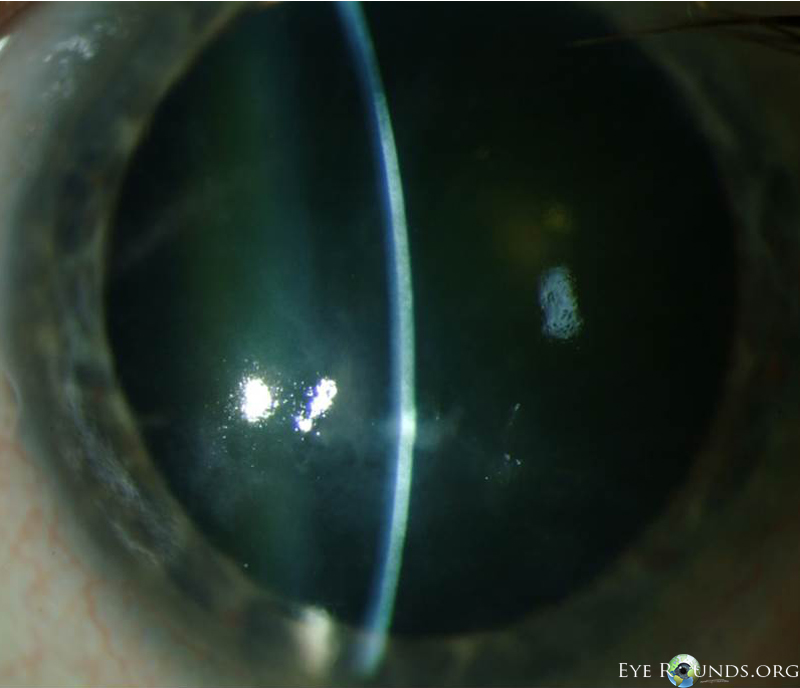
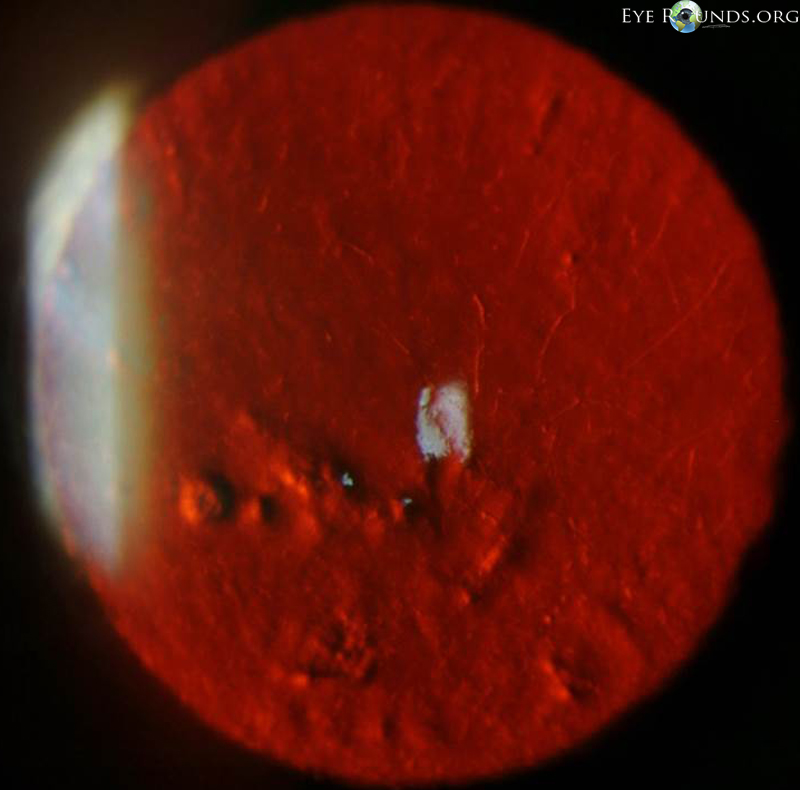
Salzmann’s nodules inferior to visual axis with diffuse anterior stromal haze all the way to the limbus, diffuse punctate epitheliopathy particularly inferiorly in an exposure pattern, reduplicated basement membrane with lattice lines |
Salzmann’s nodules inferior to visual axis with diffuse anterior stromal haze all the way to the limbus, diffuse punctate epitheliopathy particularly inferiorly in an exposure pattern, reduplicated basement membrane with lattice lines |
Anterior Chamber |
Deep and quiet |
Deep and quiet |
Iris |
Normal architecture |
Normal architecture |
Lens |
2+ nuclear sclerosis |
2+ nuclear sclerosis |
Vitreous |
Normal |
Normal |
 |
 |
 |
 |
 |
See Figure 6.
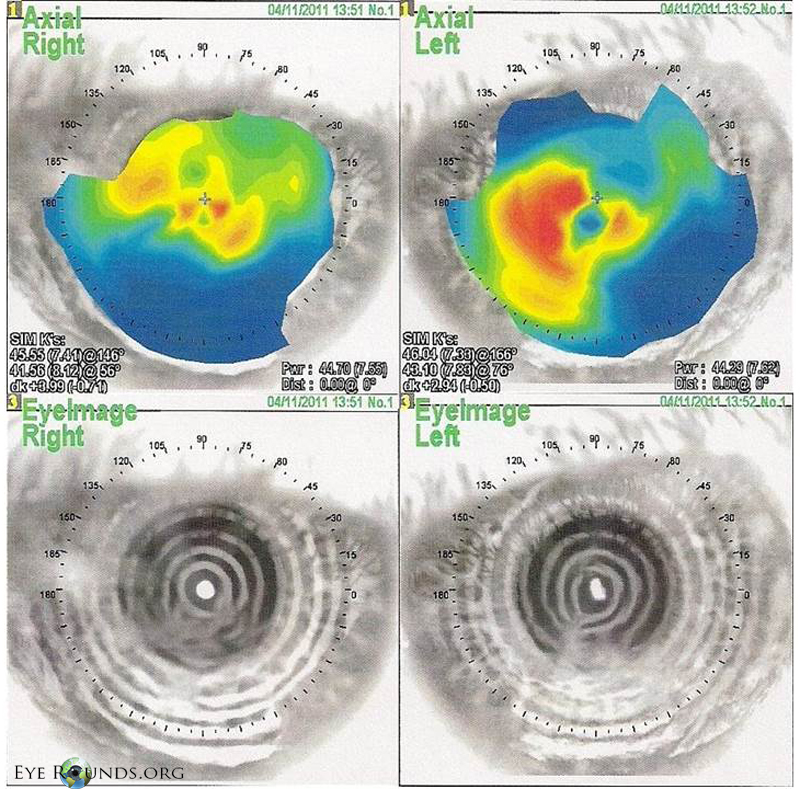 |
NIDEK corneal topography showing significant disruption of the anterior corneal surface with very irregular astigmatism and distorted placido mires.
The patient was initially evaluated by the Cornea Service at the University of Iowa and underwent superficial keratectomy (SK) of the left eye for suspected anterior corneal dystrophy. Vision improved to 20/80 (from 20/100) at post-operative week one and the patient was then referred to the Oculoplastics Service to evaluate for a possible cause of the exposure keratopathy in the setting of inferior scleral show and lid retraction.
Examination revealed a bilateral cranial nerve VII palsy (forehead weakness) and skin laxity. Given these findings and the history of similar pathology in multiple family members, Meretoja’s syndrome was suspected. Due to the bilateral cranial nerve deficits, the patient was referred to the Neuro-Ophthalmology service for further evaluation. No other cause for the deficits was identified and the diagnosis of Meretoja’s syndrome was made.
The patient’s exposure keratopathy was treated with bilateral cheek lift, enduragen graft, lateral tarsal strip, upper canthoplasty, and upper lid gold weight placement to optimize lid positioning. Unfortunately she has now developed worsening Salzmann’s nodularity limiting vision to 20/100 OU and keratectomy is being planned in the near future.
Lattice corneal dystrophy, gelsolin type, or lattice corneal dystrophy type II (LCD2) is a systemic amyloidosis with severe manifestations in the skin, cranial nerves and cornea. This was first described by ophthalmologist Jouko Meretoja in 1969 at the University of Helsinki Department of Ophthalmology in Finland after observational case reports of patients presenting with corneal lattice dystrophy and mask-like facies, and thus carries the eponym Meretoja’s Syndrome [1]. It is also known as familial amyloidosis - Finnish type, familial amyloid polyneuropathy type IV, systemic amyloidosis type V, or AGel amyloidosis. It is an autosomal dominant inherited amyloidosis that typically presents in the third decade of life with a corneal lattice dystrophy demonstrating amyloid deposition in the corneal stroma as the initial sign of disease [2].
This initial presentation may be mild, but as amyloid deposits accumulate in the cornea or other organs, typically cranial nerves and skin, morbidity increases. This commonly manifests as paralysis of the facial nerve (CN VII) and cutis laxa (elastolysis, in which the skin becomes inelastic), giving patients the characteristic facial appearance described as “hound-like” or “mask-like”. The “triple-whammy” of amyloid deposition in the cornea, exposure keratopathy, and meibomian gland obstruction secondary to orbicularis weakness may result in significant corneal pathology. Treatment should address all aspects of the disease in order to optimize ocular health.
As with other amyloid diseases, LCD2 is based on misfolded proteins forming β-sheets instead of α-helices creating fibrillar amyloid structures that accumulate in affected organs to cause pathology. LCD2 is an autosomal dominant mutation that has been mapped to chromosome 9 and has 100% penetrance with variable expressivity. The mutation is a point mutation at 9q32-34 in the gene coding for the protein gelsolin. This mutation leads to a single base substitution of adenine for guanine at position 654 that alters the amino acid sequence by replacing aspartic acid with asparigine in codon 187. The result is abnormal proteolysis and unstable protein products that have tendency to form amyloidogenic fragments [3].
Gelsolin is a protein that has two wild-type variants (products of alternate splicing) found either intracellularly or in plasma. In LCD2, both variants contain the mutation, but the protein secreted into the plasma is responsible for amyloid deposition in affected organs and the resultant pathology [3].
Post-mortem autopsy examination of LCD2 patients reveals deposition of amyloid in many ocular structures including varying levels of the cornea (ranging from just beneath Bowman’s layer to the deep stroma), conjunctiva, sclera, and ciliary body, and also the perineurium of affected nerves, walls of affected blood vessels, and basal membranes of the epidermis in affected cutaneous structures. The fact that avascular structures are affected suggests that local amyloid production mechanisms also play a role in the disease [3]. No amyloid has been found in the trabecular meshwork. The secondary chronic open-angle glaucoma associated with LDC2 is thought to be due to the mutated intracellular variant of gelsolin affecting actin in the trabecular muscle cells causing their dysfunction [3].
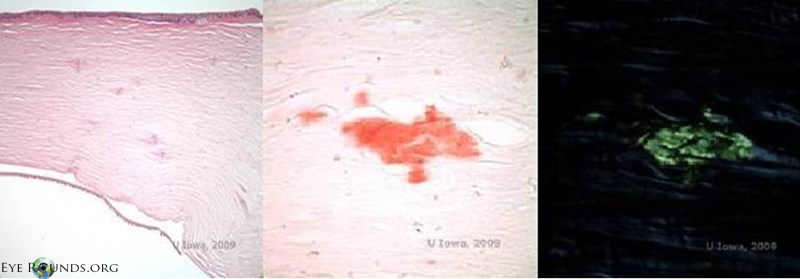
Except for DNA verification, there is no laboratory test of specific diagnostic significance for LCD2. Affected tissue that stains positive with amyloid markers on histology can aid in the clinical diagnosis (Figure 7). The diagnosis is typically clinical and dependent upon observation of lattice corneal dystrophy, a family pedigree consistent with an autosomal-dominant inheritance pattern and suggestion of other systemic features consistent with LCD2 that differentiate it from the other lattice corneal dystrophies.
 |
Lattice corneal dystrophy type II (Meretoja’s syndrome)
Signs
|
Symptoms
|
Associated Syndromes
|
Treatment
|
Kirkpatrick CA, Shah SS, Goins KM, Allen RC. Lattice Corneal Dystrophy Type II – Meretoja’s Syndrome: A 68-year-old female presents with progressive decline in vision and glare. EyeRounds.org. August 21, 2013; available from https://eyerounds.org/cases/176-Meretoja.htm.

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
